

Детектор электронного захвата — киберпедия
В основе функционирования детектора электронного захвата лежит то положение, что молекулы многих веществ способны реагировать со свободными электронами с образованием стабильных отрицательных молекулярных ионов.
Принципиальная схема детектора электронного захвата:
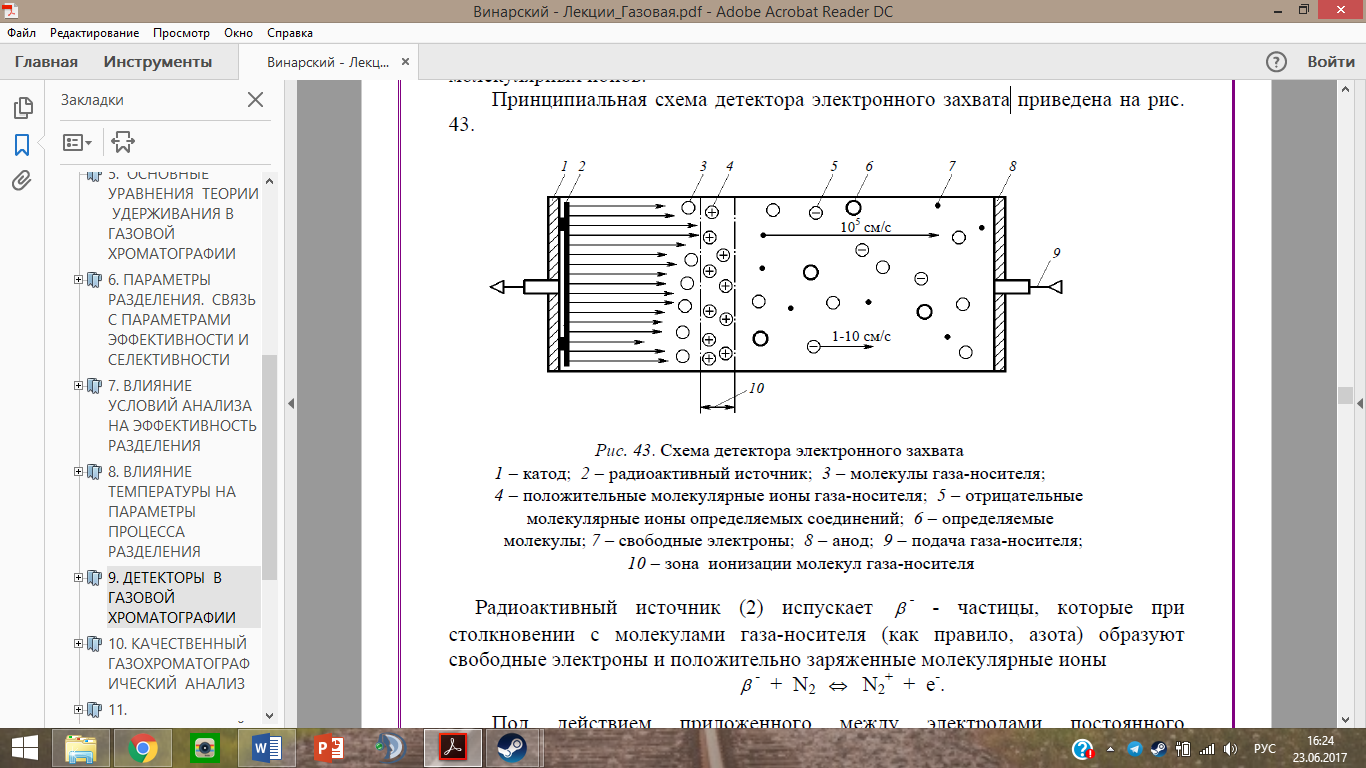
Под действием приложенного между электродами постоянного напряжения образовавшиеся в зоне ионизации свободные электроны движутся к аноду с очень высокой скоростью (порядка 105 см/c), несмотря на встречное движение потока газа-носителя. При этом в системе возникает электрический ток, который усиливается и регистрируется измерителем малых токов.
При практическом использовании детектора электронного захвата необходимо учитывать следующие его особенности:
• температура детектора должна быть несколько выше температуры термостата колонок с целью устранения конденсации пробы и неподвижной фазы в детекторе;
• для сильно захватывающих электроны веществ, следует использовать лишь небольшие количества проб. Большие по количеству пробы насыщают детектор в течение нескольких часов;
• многие органические растворители(кетоны, спирты, хлорсодержащие соединения) способны захватывать электроны. Введение большого количества таких растворителей приводит к быстрому насыщению детектора.
Детектор электронного захвата – потоковый детектор.
Детектор электронного захвата применяют для анализа:
• токсичных соединений в воздухе;
• остаточных количеств пестицидов, гербицидов, инсектицидов и некоторых других соединений, вредных для человека, в крови, в пищевых продуктах,
спиртных напитках, в биологии для анализа аминов, токсичных соединений, гормонов, канцерогенных веществ и метаболитов;
• для анализа летучих галогенсодержащих соединений в различных пробах;
• для анализа некоторых металлоорганических и неорганических соединений.
Основные недостатки детектора электронного захвата:
• чувствительность к изменению температуры;
• сравнительно невысокий верхний температурный предел использования;
• малая линейная область детектирования;
• возможность протекания следующих побочных процессов:
• возникновение пространственного заряда;
• возникновение контактных потенциалов;
• изменение энергии электронов в процессе детектирования, приводящих к искажению результатов анализа.
74. Капиллярная газовая хроматография. Устройство основных узлов газового хроматографа. Принципиальная схема. Капиллярный испаритель для введения образца с делением и без деления потока.
Хроматография – это метод разделения и определения веществ, основанный на распределении компонентов между двумя фазами – подвижной и неподвижной. Неподвижной (стационарной) фазой служит твердое пористое вещество (часто его называют сорбентом) или пленка жидкости, нанесенная на твердое вещество. Подвижная фаза представляет собой жидкость или газ, протекающий через неподвижную фазу, иногда под давлением.
Время удерживания tR — время от момента ввода анализируемой пробы до момента регистрации максимума хроматографического пика.По времени удерживания проводится идентификация разделяемых веществ.Время удерживания складывается из двух составляющих – времени пребывания веществ в подвижной фазе (tm) и времени пребывания в неподвижной фазе (ts).
Степень размывания хроматографического пика определяет эффективность колонки. Чем эффективнее колонка, тем уже пик, тем большее число компонентов можно разделить за более короткое время, т.е. время анализа сокращается. Количественно эффективность колонки может быть выражена числом теоретических тарелок N
Сопоставление площадей или высот хроматографических пиков позволяет оценить количественный состав смеси. В хроматографии используют три основных метода количественного анализа.
Метод абсолютной калибровки обычно предполагает построение градуировочного графика по стандартным смесям, как и в других физических методах.
В методе внутренней нормализации предполагается, что пики всех возможных компонентов смеси зафиксированы на хроматограмме, и сумма их площадей (S) равна 100%.
Метод внутреннего стандарта предусматривает введение в анализируемый образец известного количества эталонного соединения, хроматографирование полученной смеси.
Капиллярная хроматография – это вид хроматографии, в котором для разделения соединений используют капиллярные колонки. Сорбент (неподвижная фаза, жидкая пленка) в таких колонках расположен только на внутренних стенках капилляра, а центральная часть по сечению остается незаполненной (полые или открытые колонки). Движение газа — носителя (подвижная фаза) в колонках без наполнителя сопровождается значительно меньшими энергетическими потерями, чем в заполненных пористым материалом трубках, что повышает эффективность разделения веществ
Для приготовления капиллярных колонок используют стеклянные, кварцевые или металлические трубки, которые должны удовлетворять следующим требованиям:
– капилляр должен иметь нужную длину и постоянный диаметр по всей длине, причем эти параметры не должны изменяться под действием температуры и давления;
– внутренняя поверхность капилляра должна быть химически однородной, на ней не должно быть больших трещин и пор;
– поверхность должна адсорбировать сорбаты, жидкие неподвижные фазы и газ-носитель в минимальной степени;
– поверхность должна прочно и равномерно смачиваться неподвижной фазой, т.е. на поверхности должен быть гомогенный разделяющий слой неподвижной фазы;
– капилляры должны обладать необходимой механической прочностью.
Длина колонки 10-100 м
Внутренний диаметр 0,25-0,35 мм
Среднее число теоретических тарелок 150 000
Толщина пленки 0,005-0,5
Приготовление колонки состоит из ряда этапов:
изготовления капилляра;
подготовки внутренней поверхности капилляра – травлением или дезактивацией;
нанесения неподвижной фазы;
кондиционирования и испытания капиллярной колонки.
По сравнению с другими видами хроматографии К.х. позволяет:
а) увеличить удельную и общую эффективность разделения, а также скорость изменения температуры при ее программировании;
б) упростить сочетание газовой хроматографии с масс-спектрометрией;
в) снизить температуру хроматографической колонки и анализировать термически нестойкие соединения;
г) уменьшить расход подвижной фазы
д) (малые объемы вводимых проб для капиллярных калонок)
Существует несколько типов капиллярных колонок:
1. Капиллярные колонки с пленкой жидкой неподвижной фазой (WCOT) тонкая пленка неподвижной фазы нанесена непосредственно на внутреннюю поверхность колонки толщина пленки 0,01-1 мкм
2. Капиллярные колонки с пористым слоем, пропитанным жидкой фазой, (PLOT) на внутренних стенках расположен слой носителя, несущего неподвижную фазу толщина пленки 1 — 5 мкм
3. Капиллярные колонки с твердым носителем (SCOT) на внутренних стенках напылен слой твердого носителя толщина пленки 10 мкм
4. Капиллярные колонки с химически привитой неподвижной фазой
Особенности капиллярной хроматографии предъявляют весьма жесткие требования к детекторам. Они должны обладать высокой чувствительностью и скоростью регистрации сигнала и иметь небольшой объем измерительной камеры. В наибольшей степени удовлетворяет всем требованиям пламенно-ионизационный детектор.
Применение
Капиллярная хроматография позволяет решать целый ряд сложных аналитических задач, в том числе различать вещества, отличающиеся на 2-3 единицы молекулярной массы, проводить анализ биологических жидкостей на содержание стероидных гормонов, определять состав душистых веществ, контролировать содержание ароматических углеводородов в объектах окружающей среды.
Устройство основных узлов газового хроматографа
| устройство для ввода пробы |
| устройство регулировки давления |
| система управления прибором и обработки данных |
Капиллярный испаритель для введения образца с делением и без деления потока.
(Испарители предназначены для ввода жидких или газообразных проб с помощью шприца)
Введение с делением потока обычно применяется для анализа основных, концентрированных компонентов (при высоких концентрациях веществ в образце, и когда образцы не удаётся разбавить).
Введение без деления потока – для анализа микропримесей.
При работе в режиме с делением потока жидкий образец вводят в нагретый испаритель для введения образца 555555555555555555555555555555555555555555555555555555555555555555555555555555555555555555555555, где он быстро испаряется. При этом небольшая часть образованного пара поступает в колонку, а большая часть выходит наружу через канал сброса. Отношение протока через колонку к потоку на сброс (коэффициент деления потока) задается заранее.
При работе в режиме без деления потока клапан, управляющий сбросом, закрывается на время введения образца. Клапан остается закрытым, пока образец не будет перенесен в колонку. После завершения ввода образца клапан, управляющий сбросом, снова открывается, что дает возможность удалить из испарителя труднолетучие компоненты и подготовить его к следующему вводу пробы.
75. Капиллярная газовая хроматография. Капиллярные колонки и их характеристика. Термостат. Установка капиллярных колонок в термостат
Капиллярные колонки
Снаружи капиллярные колонки из плавленого кварца покрыты защитным полиимидным слоем, который предохраняет колонку от механических повреждений.
Различают 3 типа колонок: WCOT,SCOT, PLOT
· WCOT – неподвижная фаза представляет собой полимер (органополисилоксан или полиэтиленгликоль), нанесенный на внутреннюю поверхность капилляра.Эти колонки универсальны и наиболее часто применяются в газохроматографическом анализе.
· PLOT – содержат на внутренней поверхности капилляра тонкий слой пористого адсорбента (оксид алюминия, углеродистый материал).Колонки PLOT идеально подходят для разделения соединений, которые при комнатной температуре газообразны, для разделения реактивных газов и легколетучих полярных газов, благородных газов, легких углеводородов.
· SCOT– пористая поверхность внутренних стенок
(более подробно о колонках в 74 билете)
Термостат газового хроматографа предназначен для программированного нагревания помещенных в него колонок.Задание постоянной температуры термостата позволяет проводить хроматографический анализ в изотермическом режиме, а программирование термостата позволяет повышать температуру в процессе анализа согласно заданной температурной программе.
Программирование температуры может быть как односегментным, так и многосегментным.
При односегментном программировании температуры задаются температуры и длительности начального и конечного изотермических участков, а также скорость увеличения температуры между ними.
Многосегментное изменение температуры программируют аналогичным образом. Программируется изменение температуры от исходной до конечной, но с различными скоростями, временами выдержки и промежуточными температурами. При программировании предусмотрена возможность не только повышения, но и понижения температуры.
76. Капиллярная газовая хроматография. Типы полисилоксановых покрытий капиллярных колонок. Взаимодействие анализируемых веществ и неподвижной фазы. Характеристики селективности неподвижной фазы по Мак-Рейнольдсу.
1)Самыми распространенными НФ, используемыми для получения WCOT- колонок, являются диметилсиликоновые, фенилметилсиликоновые, смешанные фенилметилполисиликоновые цианопропилметилсиликоновые, цианопропилфенилсиликоновые, трифторпропилметилсиликоновые фазы, а также полиэтиленгликоли (карбовакс). Большинство высокоэффективных разделений в газовой хроматографии проводится с использованием этих фаз.
2)Взаимодействие анализируемого вещества и неподвижной фазы (НФ) представляет собой сумму неполярных дисперсионных и специфических полярных взаимодействий. К последним относятся диполь-дипольные взаимодействия и водородные связи.Поэтому, на основе преобладающего типа взаимодействий, предоставляемых аналиту НФ, фазы обычно подразделяют на два класса — полярные и неполярные.
3)Система для характеристики селективности НФ была предложена Мак-Рейнольдсом. Согласно этой системе, селективность оценивается как разность индексов удерживания Ковача для пяти выбранных стандартных соединений (бензол, н-бутанол, пентанон-2, нитропропан и пиридин), определенных на колонке с исследуемой НФ, и индексов удерживания Ковача, определенных на колонке со скваланом (неполярный высококипящий углеводород).
77. Капиллярная газовая хроматография. Характеристика детекторов. Пламенно-ионизационный детектор (ПИД или ДИП.). Детектор по теплопроводности (катарометр).
Детекторы предназначены для непрерывного измерения концентрации веществ на выходе из хроматографической колонки. Принцип действия детектора должен быть основан на измерении такого свойства аналитического компонента, которым не обладает подвижная фаза.
Детекторы бывают интегральные и дифференциальные
· Интегральные – регистрируют изменение во времени суммарного количества всех компонентов
· Дифференциальные – измеряют мгновенную концентрацию компонентов
1. Концентрационный детектор
2. Потоковый детектор
Пламенно – ионизационный детектор:
ДИП является детектором потокового типа, то есть его сигнал пропорционален общей массе аналита и не зависит от выбранной скорости потока газа носителя через колонку
Газ-носитель (пф) из колонки приносит компоненты образца в пламенно – ионизационный детектор. Эти компоненты попадают в пламя, получаемое при сжигании водорода с воздухом. При сгорании органических соединений в пламени образуются СНО* — радикалы.При их взаимодействии с молекулами воды в пламени генерируются положительно заряженные ионы (преимущественно, Н3О ).Эти ионы под действием постоянного напряжения движутся к коллектору, расположенному над горелкой детектора. При отсутствии компонентов образца число образуемых в пламени ионов мало. При сжигании органических веществ количество ионов увеличивается. Ток регистрируется электрометром, сигнал преобразуется в цифровой вид и направляется в систему управления прибором и обработки данных.
CnHm O → n CHO e—
CHO nH2O → (H2O)nH CO
§
• Если температура детектора составляет менее 150 ºС
• Если перекрыты потоки воздуха или водорода
• Если попытка автоматического поджига оказалась неудачной
Детектор по теплопроводности (Катарометр):
Это детектор концентрационного типа, сигнал которого зависит не только от массы (концентрации аналита), но и от времени нахождения аналита в ячейке детектора, то есть от суммарной скорости потока газа-носителя и поддувочного газов через ячейку.
(измерение теплопроводности газа-носителя в присутствии других веществ)
Детектор измеряет различие в теплопроводности чистого газа – носителя (газ-сравнения) и смеси газа-носителя с определяемым веществом (газ-колонки).
В детекторе имеется нить (вольфрамовая), нагреваемая с помощью электрического тока до температуры, превышающей температуру корпуса детектора. Через нить пропускаются чередующиеся потоки газа сравнения и газа колонки. Потоки газа, проходящие через нить, переключаются пять раз в секунду. Температура нити поддерживается постоянной, а необходимая для поддержания этой температуры мощности измеряется и регистрируется. При добавлении испытываемой пробы мощность, требуемая для поддержания температуры нити постоянной, изменяется.
78. Капиллярная газовая хроматография. Эффективность колонок и число теоретических тарелок. Параметр хроматографического разделения.
Время удерживания tR — время от момента ввода анализируемой пробы до момента реОно складывается из двух составляющих – времени пребывания веществ в подвижной фазе (tR0) и времени пребывания в неподвижной фазе (tR‘):
Значение tR зависит от природы вещества и колонки (тип покрытия, толщина покрытия для капиллярных колонок, плотности набивки колонки для набивных), поэтому даже на аналогичных колонках оно различается.
Для характеристики истинной удерживающей способности колонки вводят исправленное время удерживания – tR‘ (из времени удерживания вычесть время пребывания в подвижной фазе)
Эффективность колонок и число теоретических тарелок
Теоретическая тарелка – это условный участок хроматографической колонки, в пределах которого устанавливается равновесие вещества, распределенного между подвижной и неподвижной фазами (одна теоретическая тарелка отвечает однократному распределению между фазами).Чем больше теоретических тарелок в колонке, чем большее число раз устанавливается равновесие, тем эффективнее колонка.
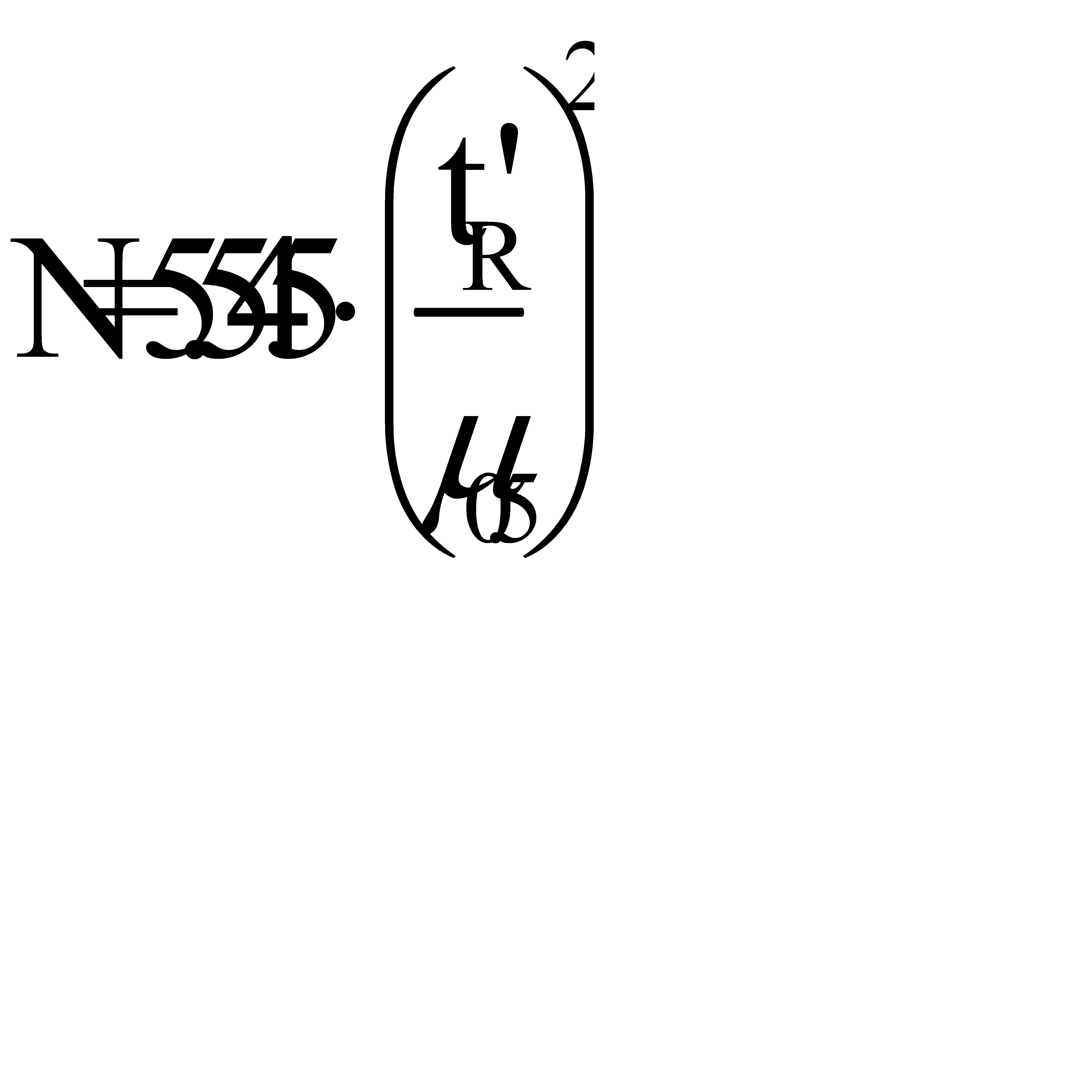
Эффективность колонки характеризуется числом теоретических тарелок и высотой, эквивалентной теоретической тарелке (ВЭТТ). Число теоретических тарелок,обозначаемое как n,является основной характеристикой и рассчитывается по формулам:
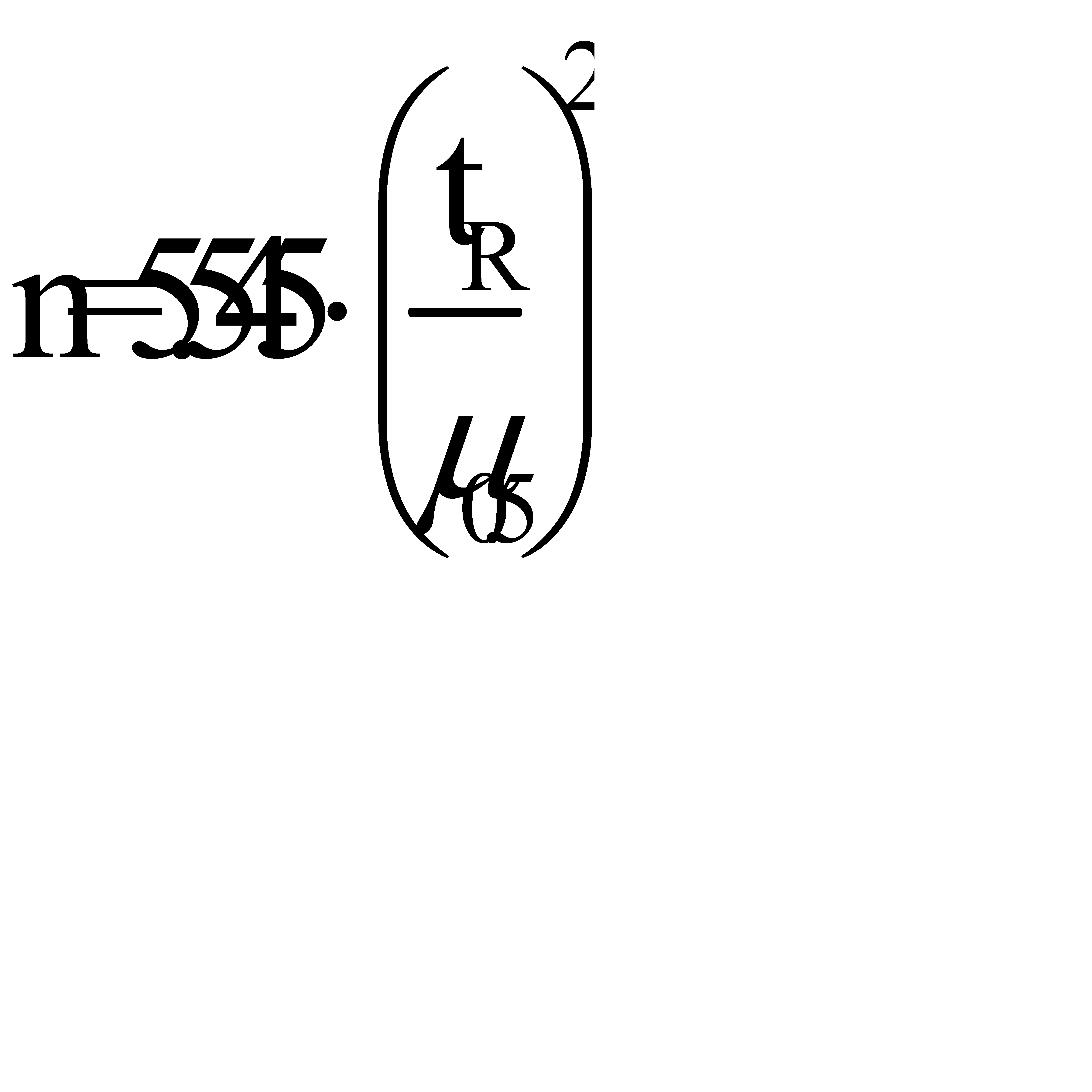 и
и 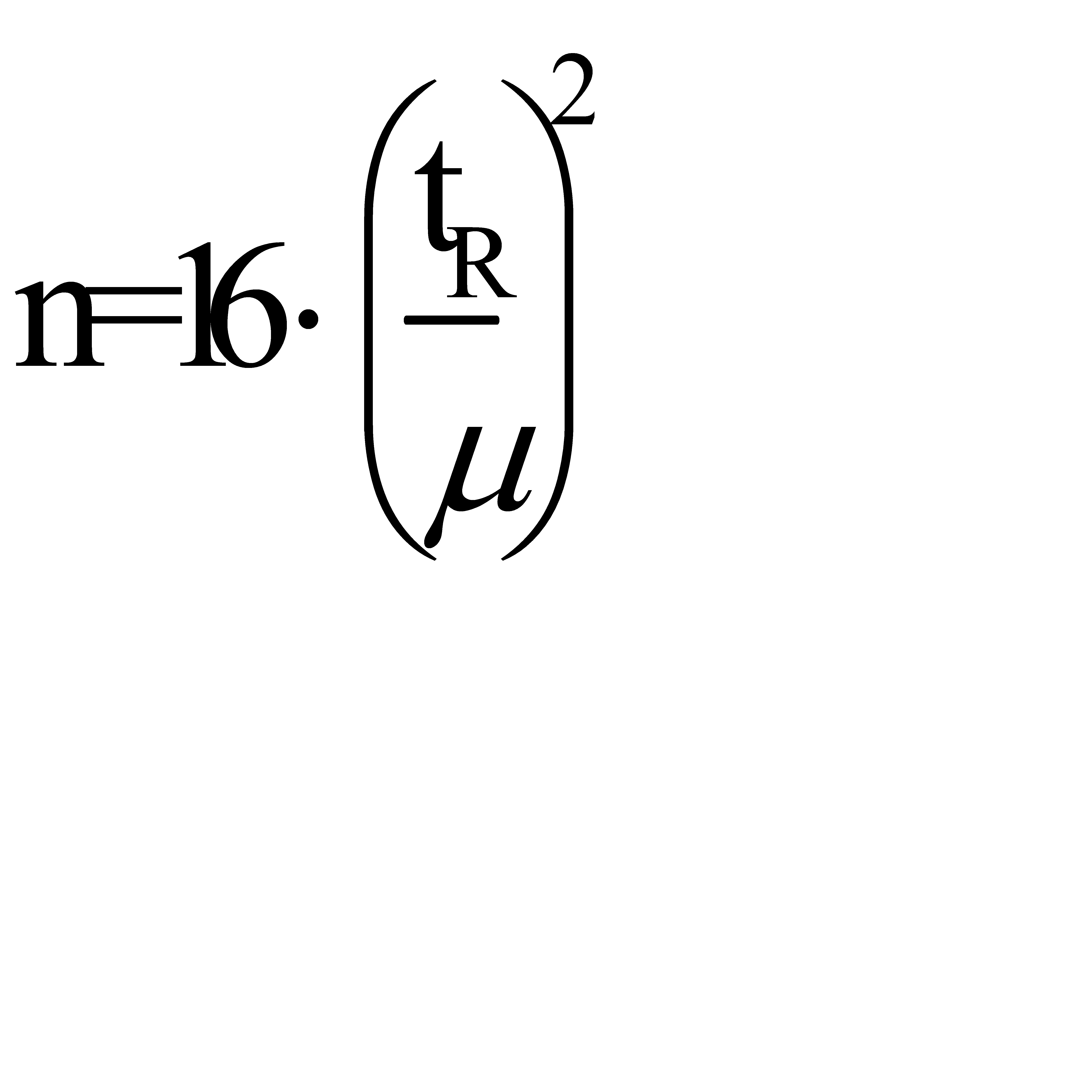 где μ и μ0,5 соответственно ширина пика у основания и на середине высоты, измеренные в единицах времени удерживания.
где μ и μ0,5 соответственно ширина пика у основания и на середине высоты, измеренные в единицах времени удерживания.
Высота, эквивалентная теоретической тарелке (Н), рассчитывается по формуле, где L – длина колонки, и выражается в см.
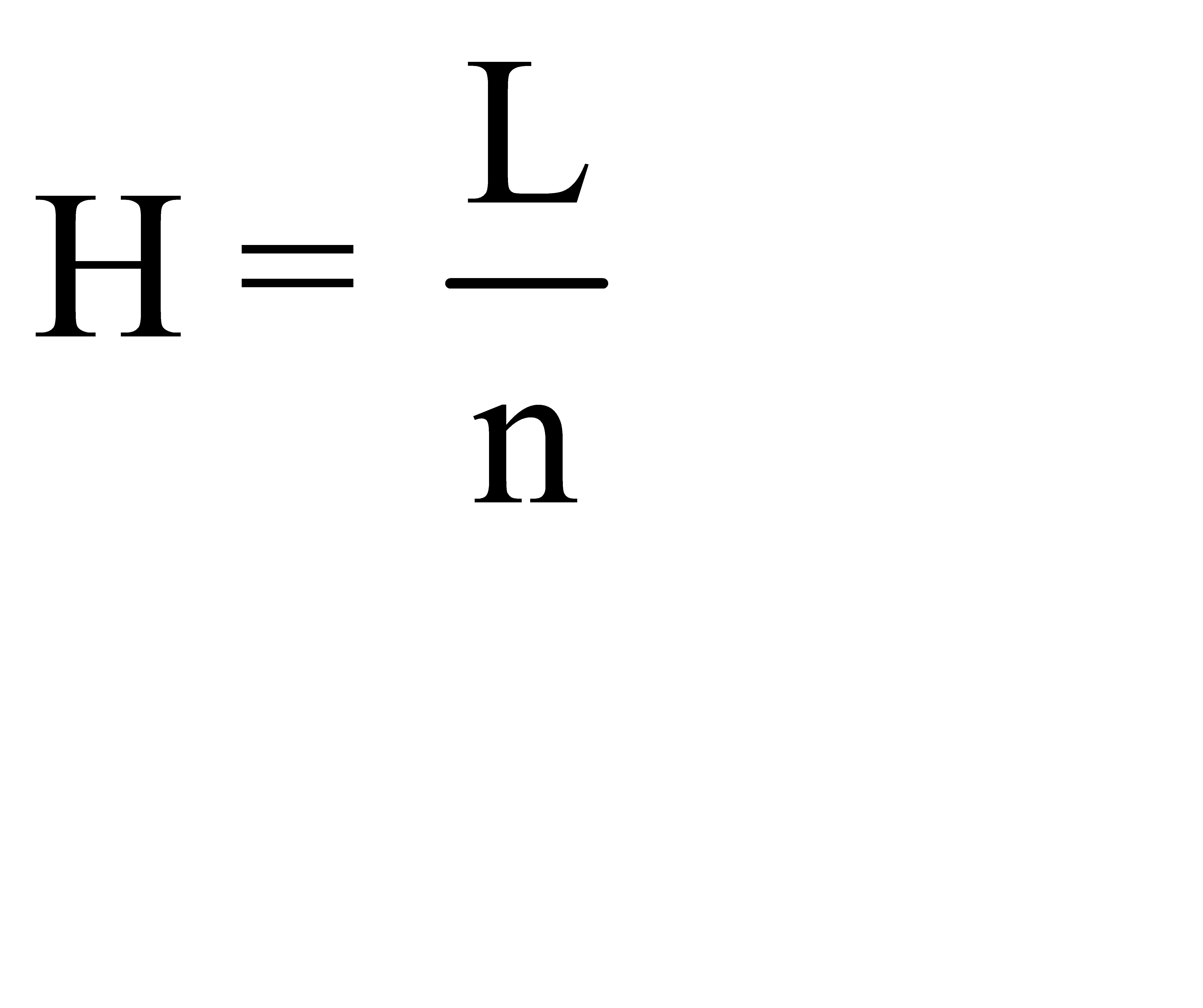
Эффективность колонки тем выше, чем меньше высота, эквивалентная теоретической тарелке, и больше число теоретических тарелок.
Число эффективных теоретических тарелокN рассчитывают для учета влияния коэффициента емкости колонки (коэффициент емкости характеризует емкость удерживания колонки и показывает, во сколько раз вещество дольше находится в неподвижной фазе, чем в подвижной)

Разделение двух соседних пиков характеризуется разрешением Rs.
Разрешение является мерой полноты разделения двух веществ. Разделение считается полным, если RS> 1,5
79. Капиллярная газовая хроматография. Определение количественного состава смеси методом внутренней нормализации с использованием калибровочных коэффициентов
Предполагается, что пики всех возможных компонентов смеси зафиксированы на хроматограмме, и сумма их площадей (S) равна 100% (содержание компонента в смеси равно отношению сигнала этого компонента к сумме сигналов всех компонентов, при этом могут быть использованы соответствующие калибровочные коэффициенты, на которые умножают площади пиков перед расчетом).
Объем пробы, вводимой в хроматограф, в этом случае не имеет значения, т.е. точная дозировка пробы в данном методе не требуется. Метод внутренней нормализации широко применяется в хроматографическом анализе многокомпонентных смесей; обычно им пользуются, если требуется узнать полный состав смеси
Метод внутренней нормализации дает правильные результаты только при выполнении следующих условий:
· проба полностью испаряется в дозирующем устройстве;
· все компоненты пробы полностью элюируются из разделительной колонки;
· все компоненты регистрируются детектором;
· пики на хроматограмме хорошо разрешены;
· все компоненты пробы заранее идентифицированы, то есть известно, какому соединению соответствует каждый пик на хроматограмме;
· для всех компонентов известны специфические поправочные коэффициенты, учитывающие различную чувствительность детектора к разным веществам,
· сигнал детектора линеен во всем диапазоне концентраций компонентов.
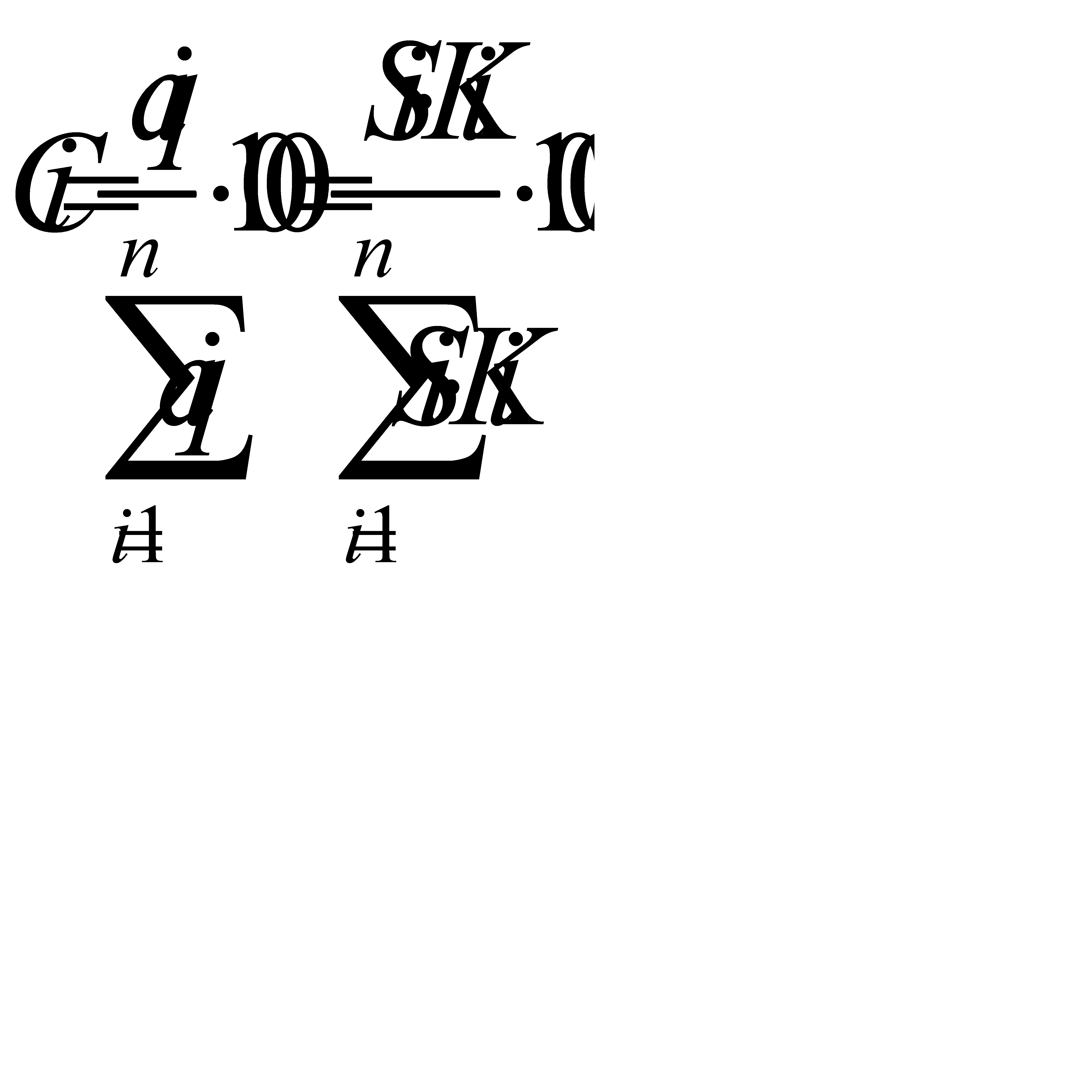
где Si – параметр хроматографического пика (площадь или высота); Ki – поправочные коэффициенты, зависящие от относительной чувствительности детектора к разным компонентам пробы.
80. Капиллярная газовая хроматография. Определение содержания компонента методом внешнего стандарта (или методом абсолютной градуировки). Расчеты по методу внешнего стандарта.
Метод абсолютной градуировки (метод внешнего стандарта) основан на использовании зависимости площади (S) хроматографического пика (или его высоты h) от концентрации соответствующего вещества в смеси (Сi) при постоянном объеме вводимой пробы, или от массы соответствующего вещества в пробе (q), введенной в хроматограф. Такую зависимость называют градуировочной.
Зависимость определяют экспериментально, разделяя приготовленные смеси известного состава, и выражают способами:
а) с помощью пропорции, если производят сравнение с одним эталоном. Такой упрощенный метод внешнего стандарта часто используют в фармацевтике, при анализе проб, имеющих близкий диапазон концентраций.
Готовят и анализируют один эталонный раствор, рассчитывая градуировочный коэффициент:
Кст =Сст / Sст. (3.4)
Затем хроматографируют анализируемый раствор, определяют площадь пика анализируемого соединения Siи рассчитывают концентрацию:
Сi = Кст· Si. =Si ·Сст/ Sст
б) графически, в координатах S – q или S – Сi. При этом неизвестную концентрацию (или массу), соответствующую измеренной площади хроматографического пика, находят по графику.
в) либо используют уравнение, описывающее графическую зависимостьСi= Кi · Si, где Кi -градуировочный коэффициент, Si— площадь пика, Сi— концентрация вещества i
Условия хроматографирования при градуировке и определении содержания компонентов пробы должны быть идентичными. Одним из основных условий получения точных результатов по методу абсолютной градуировки является воспроизводимость размера пробы, поэтому метод, как правило, используют при наличии автоматических дозирующих устройств.
Воспроизводимость и правильность результатов анализа зависят от тщательности приготовления эталонных смесей и от постоянства режима работы хроматографической аппаратуры. Поскольку чувствительность детектора к различным веществам неодинакова, необходимо нахождение калибровочных коэффициентов для каждого индивидуального вещества. При переходе к другим условиям детектирования или даже к другим условиям разделения смеси коэффициенты определяют заново.
Таким образом, недостатком метода абсолютной калибровки является длительность предварительной подготовки к выполнению анализов. Поэтому метод используется в основном в тех случаях, когда проводятся серийные анализы однотипных проб по неизменной методике, причем надо определять не все, а лишь некоторые компоненты пробы.
81. Капиллярная газовая хроматография. Количественные характеристики аналитического метода. Предел обнаружения. Нижняя граница определяемых содержаний.
Для характеристики возможностей метода применительно к КАЧЕСТВЕННОМУ АНАЛИЗУ обычно приводят такую характеристику как предел обнаружения (Смин илиSDL ).
Для расчета предела обнаружения аналитов измеряют и статистически обрабатываютаналитический сигнал фона(Sblank). Определяют величину его стандартного отклонения относительно среднего значения (s0 или δ).
Предел обнаружения (SDL) определяют путем экспериментального измерения дисперсии уровня аналитического сигнал фона(Sblank) по 3δ — критерию (или, иногда, 2δ-критерию), где δ- стандартное отклонение.
Нередко аналитический сигнал фона (Sblank) обусловлен не только также шумами, возникающими в измерительных приборах, усилителях, но и наличием примесей определяемого компонента в реагентах, растворителях, в матрице образца, и т.п.
В таком случае аналитический сигнал фона не равен нулю, и его измеряют отдельно для каждого аналита, как площадь пика на хроматограмме растворителя (Signal blank) при временах удерживания, соответствующих аналитам:
S blank = S (tRanalyte)
При измеренном аналитическом сигнале фона полезным сигналом(S) при хроматографировании пробы необходимо считать разницу между измеренным сигналом (Smeasured) и аналитическим сигналом фона (Sblank):
S = Smeasured — Sblank
В зависимости от того, вычитают или нет аналитический сигнал фона (Sblank) из измеренного, для измерения сигнала, соответстветствующий пределу обнаруженияаналита используют формулы:
1)полезный аналитический сигнал, соответстветствующий пределу обнаруженияаналита определяют по формуле : SDL = 3 s0
2)аналитический сигнал, соответствующий пределу обнаруженияаналита определяют по формуле SDL = Sblank 3 s0
(при этом аналитический сигнал фона (Sblank) не вычитают из измеренного сигнала !)
СDL определяют по градуировочному графику на основе рассчитанных величин SDL.
Для характеристики возможностей метода применительно к КОЛИЧЕСТВЕННОМУ АНАЛИЗУ обычно приводят диапазон определяемых содержаний– область значений определяемых содержаний, ограниченную верхней и нижней границей определяемых соединений.
Верхняя граница (св) — наибольшее значение концентрации или количества компонента, определяемое по методике. Оно, как правило, ограничено либо изученным интервалом, либо возможностью измерения сигнала с достаточной точностью. Может быть легко увеличено путем разбавления исходного раствора.
Нижняя граница (сн) — наименьшее содержание, определяемое по методике.
Один из способов определения нижней границы определяемыхсодержаний состоит в определении того минимального количества, которое можно измерить с относительным стандартным отклонением sr< 0.33.
Другой способ состоит в измерении предела количественного определенияСLOQ. Для измерении предела количественного определения проводят измерение величины аналитического сигнала фона (Sblank) и дисперсии уровняаналитического сигнала фона(δ).
Аналогично определению SDL, для расчетов можно использовать измеренный или полезный аналитический сигналы. Сигнал, соответствующий пределу количественного определения (SLOQ), определяют по 10 δ — критерию, где δ — стандартное отклонение аналитического сигнала фона.
Аналитический сигнал (где сигнал — измеренная величина), соответствующий пределу количественного определения аналита (SLOQ) определяют по формуле 4.14. Полезный аналитический сигнал (где сигнал — измеренная величина минус Sblank), соответствующий пределу количественного определения аналита (SLOQ) определяют по формуле 4.15.:
SLOQ = Sblank 10 s0 (SLOQ — это измеренный аналитический сигнал) (4.14)
SLOQ = 10 s0 (SLOQ — это полезный аналитический сигнал) (4.15)
Используя полученные величины SLOQ, определяют СLOQ по градуировочному графику.
82. Область применения видимой и УФ-спектроскопии в фармации: качественный и количественный анализ вещества. Исследование структурных и динамических свойств молекулярных систем.
Спектроскопия изучает взаимодействие электромагнитного излучения с веществом.
Одним из общих свойств молекул является способность к избирательному поглощению электромагнитного излучения, что и положено в основу исследования строения и идентификации веществ
Ультрафиолетовая область спектра (200-400нм)
Видимая область спектра (400-800нм)
Уменьшение интенсивности монохроматического излучения, проходящего через гомогенную поглощающую среду, количественно описывается законом Бугера-Ламберта-Бера:
log10(1/Т) = А = ε ∙ c ∙ b ,(1)
где:
- Т – пропускание, отношение интенсивности светового потока, прошедшего через вещество, к интенсивности падающего на вещество светового потока; Т = I/I0;
- I – интенсивность прошедшего монохроматического излучения;
- I0 – интенсивность падающего монохроматического излучения;
- ε – молярный показатель поглощения;
- с – молярная концентрация вещества в растворе;
- b – длина оптического пути или толщина слоя, в сантиметрах.
Величина log10(1/Т) носит название оптической плотности, обозначается буквой А и является измеряемой величиной. В отсутствии других физико-химических факторов измеренная оптическая плотность (А) пропорциональна концентрации вещества в растворе (с) и толщине слоя (b).
Величина  представляет собой удельный показатель поглощения, т.е. оптическую плотность раствора вещества с концентрацией 10 г/л (1 г/100 мл) в кювете с толщиной слоя 1 см. Величины
представляет собой удельный показатель поглощения, т.е. оптическую плотность раствора вещества с концентрацией 10 г/л (1 г/100 мл) в кювете с толщиной слоя 1 см. Величины 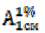 и ε связаны соотношением:
и ε связаны соотношением:

где:
М.м. – молекулярная масса исследуемого вещества.
§
Если нет других указаний в фармакопейной статье, измерение оптической плотности проводят при указанной длине волны с использованием кювет с толщиной слоя 1 см и при температуре (20 ± 1) °С по сравнению с тем же растворителем или той же смесью растворителей, в которой растворено вещество. При измерении оптической плотности раствора при данной длине волны оптическая плотность кюветы с растворителем, измеренная против воздуха при той же длине волны, не должна превышать 0,9 и, желательно, чтобы она была не менее 0,2.
Спектр поглощения представляют таким образом, чтобы оптическая плотность или ее некоторая функция были приведены по оси ординат, а длина волны или некоторая функция длины волны – по оси абсцисс.
Идентификация
Абсорбционную спектрофотометрию в ультрафиолетовой и видимой областях спектра применяют для определения подлинности лекарственных средств путем:
— сравнения спектров поглощения испытуемого раствора и раствора стандартного образца; в указанной области спектра должно наблюдаться совпадение положений максимумов, минимумов, плеч и точек перегиба;
— указания положений максимумов, минимумов, плеч и точек перегиба спектра поглощения испытуемого раствора; расхождение между наблюдаемыми и указанными длинами волн в максимумах и минимумах поглощения не должно обычно превышать ± 2 нм.
УФ-спектрофотометрия используется при установлении подлинности (идентификации), доброкачественности, количественном определении, как индивидуальных веществ, так и компонентов лекарственных форм; испытании по тестам “Растворение” и “Однородность дозирования”.
Метод применяется на таких стадиях изучения лекарственных веществ и лекарственных форм как фармакокинетика, биодоступность, изучение стабильности и установление сроков годности.
В видимой области спектра электромагнитное излучение (360-780нм) обычно поглощают окрашенные вещества: либо за счёт собственной окраски (например, рибофлавин), либо за счёт окрашенных продуктов реакции определяемых веществ с реагентами (функциональный анализ). Основная сложность при реализации методики заключается в том, что многие реакции протекают достаточно медленно (при комнатной температуре иногда часами). Увеличение температуры приводит не только к ускорению основной реакции, но и побочных.
Количественное определение
При количественном определении в УФ-области спектра точную массу или объем анализируемого образца (субстанция, таблетки, инъекционные растворы и т.д.) растворяют в подходящем растворителе, при необходимости готовят соответствующее разведение и измеряют оптическую плотность приготовленного раствора при длине волны, указанной в НД, на приборе спектрофотометре.
Определение концентрации веществ спектрофотометрическим методом основано на использовании закона Бугера-Ламберта-Бера:
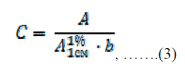
где:
С – концентрация вещества в г/100 мл;
А – оптическая плотность испытуемого раствора;
b – длина оптического пути или толщина слоя, в сантиметрах.
Применение электронной спектроскопии в ультрафиолетовой и видимой областях спектра для анализа функциональных групп ограничено тем, что соответствующие спектры имеют небольшое число полос поглощения. Их преимущество в сравнении со спектроскопией в ИК-области спектра заключается в большей чувствительности, что позволяет исследовать очень разбавленные растворы. Имеющиеся недорогие, простые в обращении современные записывающие спектрофотометры дают возможность быстро обнаруживать присутствие сильных хромофорных групп в образце.
83. Спектроскопия отражения. Спектры отражения. Сфера применения спектров внешнего и внутреннего отражения. Интенсивность спектров. Примеры.
СПЕКТРОСКОПИЯ ОТРАЖЕНИЯ — раздел спектроскопии, изучающий закономерности отражения электромагнитного излучения от различных сред. Лежит в основе методов исследования в-в по спектрам отражения.
В традиционной инфракрасной спектроскопии исследуется спектр излучения, прошедшего через образец. Существуют также методы исследования инфракрасного излучения, отражённого от поверхности образца. Они основаны на изучении:
- нарушенного полного внутреннего отражения (НПВО);
- зеркального отражения;
- скользящего отражения;
- диффузного отражения.
Существенным преимуществом таких методов является то, что удаётся изучать образцы, непрозрачные для ИК-излучения, а также обходиться без процесса пробоподготовки и проводить анализ непосредственно в полевых условиях. Кроме того, такие анализы не являются разрушающими
Спектроскопия НПВО:
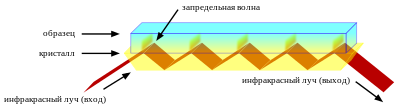
Метод основан на отражении пучка на границе раздела двух фаз: фазы кристалла НПВО с относительно высоким показателем преломления и фазы исследуемого образца с более низким показателем преломления. Если пучок излучения падает на плоскость образца под углом падения больше критического, то наблюдается практически полное отражение пучка от поверхности образца. На самом деле излучение на небольшую глубину проникает в фазу образца, где частично поглощается. При последующих попаданиях того же пучка света на образец это явление повторяется, и в результате получается некое подобие спектра поглощения. Наблюдаемые частоты поглощённого излучения будут совпадать с частотами, получаемыми в ИК-спектроскопии пропускания
Для проведения спектроскопии НПВО инфракрасные спектрометры оборудуются специальной приставкой. В ней анализируемое вещество помещается в непосредственный контакт с кристаллом и фиксируется при помощи прижимного устройства. Далее через кристалл под специально подобранным углом подаётся инфракрасное излучение, интенсивность которого фиксируется на выходе из кристалла. Обычно в дисперсионных приборах осуществляется примерно 25 отражений, а в спектрометрах с преобразованием Фурье — около шести[26].
Спектроскопия НПВО позволяет анализировать как обычные жидкие образцы, так и «сложные», например, водные растворы, пасты и гели. Поскольку кристалл НПВО легко извлекается из кюветы, нанесение и удаление образца не представляет особой трудности. Также анализу поддаются порошки и полимеры, которые прижимаются к кристаллу специальным устройством. Существуют специальные кюветы для анализа кожи, которые находят применение при изучении действия косметики и лекарств на кожу человека
Спектроскопия внешнего отражения:
Регистрируемым параметром в инфракрасной спектроскопии внешнего отражения является интенсивность отражённого света. Если разделить это значение на интенсивность падающего излучения, получится величина, называемая коэффициентом отражения. График зависимости коэффициента отражения от длины волны (или частоты излучения) содержит ту же информацию, что и классические ИК-спектры пропускания
Спектроскопия зеркального отражения:
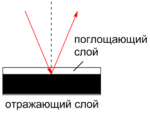
применяется для материалов, нанесённых на отражающие металлические поверхности или поверхности из другого материала, который отражает инфракрасное излучение. Суть метода заключается в том, что пучок излучения из ИК-спектрометра подаётся на изучаемую поверхность, где он проникает сквозь покрытие, отражается от подложки, снова проходит через покрытие и попадает на детектор прибора. Двойное прохождение через материал покрытия приводит к частичному поглощению ИК-излучения, что и даёт спектр поглощения для данного материала. При этом, в отличие от метода НПВО, образец может иметь шероховатую поверхность и не контактирует с кристаллом. Анализу поддаются покрытия толщиной от 1 до 100 мкм
Спектроскопию скользящего отражения применяют для изучения очень тонких слоёв на отражающей поверхности. При подаче излучения под очень большим углом падения оптический путь через слой материала сильно возрастает, что и даёт возможность получать спектры поглощения таких материалов. Если в качестве отражателя выступает вода, то этим методом можно изучать мономолекулярные слои масел, жиров, липидов и т. д. на её поверхности, при этом получая информацию о строении и плёнок. Подобным образом исследуют биологические мембраны в естественных условиях
Диффузное отражение возникает на шероховатой поверхности и не сфокусировано в определённой точке, поэтому для работы с ним используются эллипсоидные зеркала, одно из которых фокусирует ИК-излучение на образце, а второе «собирает» отражённый свет и отправляет его на детектор. Спектроскопия диффузного отражения нашла применение в анализе порошков.
Недостатком методов, использующих внешнее отражение, является сложность получаемых спектров. Обычные спектры пропускания несут в себе информацию лишь о коэффициенте экстинкции при той или иной длине волны, в то время как в спектроскопии отражения интенсивность отражённого света зависит также от коэффициента преломления. В дополнение ко всему необходимо учитывать коэффициент поглощения отражающей поверхности.
84. Молекулярные колебания. Нормальные колебания. Колебательные переходы. Инфракрасная спектроскопия. Теоретические основы метода. Аппаратура для ИК-спектроскопии. Область применения и особенности ИК-спектров вещества.
Инфракрасная спектроскопия (колебательная спектроскопия) — раздел спектроскопии, изучающий взаимодействие инфракрасного излучения с веществами.
Она занимается, главным образом, изучением молекулярных спектров испускания, поглощения и отражения, так как в ИК области расположено большинство колебательных и вращательных спектров молекул.
При поглощении излучения в ИК области колебания молекулы переходят из нулевого колебательного состояния преимущественно в первое возбужденное состояние. Такие типы колебаний называютосновными, собственными или нормальными колебаниями.
Молекулярные колебания — один из трёх типов молекулярного движения, к которым относятся также трансляционное движение (когда все атомы молекулы смещаются в одном направлении) и вращательное движение (когда молекула поворачивается на определённый угол). В отличие от последних двух случаев, когда геометрия молекулы не меняется, при колебаниях происходит изменение положения атомов относительно друг друга.
Колебательные переходы лежат в области близкого и среднего ИК излучения. Исследование колебательных спектров показывают, что их частота соответствует колебаниям отдельных атомов или групп атомов в молекуле. Эти характеристические частоты нашли широкое применение в ИК — спектроскопии для качественного анализа и изучения строения молекулы.
инфракрасный спектр — функция интенсивности пропущенного инфракрасного излучения от его частоты.
Инфракрасные спектры (колебательные спектры) (ИК-спектры) возникают вследствие поглощения энергии электромагнитного излучения при колебаниях ядер атомов в молекулах или ионах, которые сопровождаются изменением дипольных моментов, и представляют собой зависимость пропускания или поглощения от длины волны (λ) или частоты колебаний (ν).
Под инфракрасной областью (ИК-область) подразумевают электромагнитное излучение в области длин волн от 0,78 до 400 мкм. Область от 780 до 2500 нм (от 0,78 до 2,5 мкм) рассматривается как ближняя ИК-область, область от 2,5 до 25 мкм (от 4000 до 400 см-1) относится к средней ИК-области спектра и область от 25 до 400 мкм относится к дальней ИК-области. Наиболее часто используется средняя ИК-область.
Длину волны (λ) в ИК-спектрах обычно измеряют в микрометрах (микронах), мкм.
Поскольку частота колебаний в ИК-спектрах имеет большие числовые значения, обычно используют не частоты (ν), а волновые числа ( 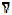 ), которые измеряются в см-1 и связаны с частотой (ν) уравнением:
), которые измеряются в см-1 и связаны с частотой (ν) уравнением:
= ν /с , где
ν – частота, Гц (с-1);
с – скорость света в вакууме, см∙с-1.
Волновое число ( ) связано с длиной волны (λ, мкм) соотношением:
= 104/λ.
Приборы
Могут быть использованы инфракрасные спектрофотометры, снабженные оптической системой (призмы или дифракционные решетки), выделяющей монохроматическое излучение в измеряемой области, или спектрофотометры с Фурье-преобразованием. В последних используется полихроматическое излучение и рассчитывается спектр в заданной области частот путем Фурье-преобразования исходных данных. В таких приборах вместо диспергирующего прибора используется интерферометр, а обработка спектральных данных производится с помощью компьютера.
(РегистрацияИК-спектров основана на тепловом действии ИК излучения, поэтому требуется оптика из прозрачных в этой области солей щелочных или щелочноземельных металлов. Анализируемые образцы или таблетируют с такой солью, или готовят неводныерастворы и помещают в специальные кюветы с окнами из NaCl, LiF, CaF2или KBr.
В методе ИК-спектроскопии наиболее широкое распространение получило исследование ИК-спектров поглощения, возникающих при прохождении ИК-излучения через вещество. Каждое вещество имеет свой колебательный спектр. Число полос поглощения в спектре, ширина, форма, интенсивность определяются структурой и химическим составом вещества. Это дает возможность по ИК-спектрам проводить качественный и количественный анализы вещества во всех агрегатных состояниях. Количественный анализ по молекулярным спектрам поглощения основан на использовании закона Бугера-Ламберта-Бера)
Качественный анализ
ИК спектроскопия является такой же специфической характеристикой, как отпечатки пальцев человека. По спектрам вещество может быть идентифицировано, если его спектр известен.
Для проведения качественного анализа проб по ИК спектрам необходимо провести его интерпретацию. При этом необходимо сочетание экспериментальных данных с теоретическим расчетом. Изучение ИК спектров веществ в настоящее время проводится двумя методами:
- выявлением характеристических частот;
- сравнением спектров сложных веществ со спектрами индивидуальных соединений.
Метод характеристических частот.Молекулы, имеющие одни и те же химические группы, часто имеют одинаковые частоты в спектре. Эти частоты называют характеристическими.
Расшифровка инфракрасного спектра производится следующим образом:
- идентификацию полос поглощения начинают с наиболее сильных и высокочастотных полос в области валентных колебаний ОН-связи.
- По таблицам характеристических частот полосу поглощения относят к колебанию конкретной связи.
- Наличие той или иной связи подтверждают деформационной полосой поглощения, относящейся к данной связи.
Метод сравнения.Идентификация неизвестного соединения по ИК спектру осуществляется сравнением его спектра с эталонными спектрами. Для этого необходима обширная картотека эталонных спектров; при этом важнейшим фактором является стандартность условий их регистрации. В настоящее время имеются многочисленные атласы органических и неорганических соединений.Идентификация веществ по ИК спектру является полностью достоверной только при точном совпаденииизучаемого спектра со спектром эталона по положению (частоте), форме и относительной интенсивности всех полос, то есть всей спектральной кривой.
Область применения
Поскольку поглощение в ИК-области связано с молекулярными колебаниями, соответственно ИК-спектры дают информацию остроении соединений. Этот метод может быть использован для анализа почти всех молекул с ковалентными связями. Если двухатомная молекула состоит из одинаковых атомов и не имеет дипольного момента, она не способна поглощать ИК-излучение (Cl2,O2 и т.п.). У многоатомных неполярных молекул в ИК-спектре активны лишь те колебания, при которых возникает периодически меняющийся дипольный момент. Остальные колебания могут проявиться в электронном спектре.
ИК-спектры могут служить источником подробной информации о структуре молекулярных соединений различной природы — витаминов, аминокислот, сложных эфиров, сахаров, спиртов и других веществ, поэтому широко используются для контроля качества. Метод ИК спектроскопии позволяет определить состояние воды в минерале, характер изоморфных примесей, степень структурной упорядоченности, отнесение минералов к определённому структурному типу и др.
ИК-спектроскопия позволяет идентифицировать почти все функциональные группы и с успехом применяется для качественного исследования органических соединений.
К недостатку этого метода следует отнести возможность исследования, как правило, лишь неводных растворов образцов, а также применение более сложной и дорогостоящей аппаратуры по сравнению с используемой в методе УФ-спектрофотометрии.
85. ВЭЖХ. Сущность хроматографического процесса. Назначение подвижной и неподвижной фазы, их природа и характеристики в методе ВЭЖХ. Классификация методов ВЭЖХ по агрегатному состоянию фаз и по способу хроматографирования. Преимущества проявительного (элюентного) анализа.
Высокоэффективная жидкостная хроматография (жидкостная хроматография высокого давления) – это метод колоночной хроматографии, в котором подвижной фазой служит жидкость, движущаяся через хроматографическую колонку, заполненную неподвижной фазой (сорбентом). Колонки для высокоэффективной жидкостной хроматографии характеризуются высоким гидравлическим сопротивлением на входе.
Основными требованиями в методе является растворимость исследуемых веществ в подвижной фазе, а также свойство удерживаться неподвижной фазы
Принцип анализа в ВЭЖХ.Анализируемую смесь растворяют в подвижной фазе и с помощью дозатора или микрошприца вводят в специальное устройство прибора. Туда же подается под определенным давлением и с определенной скоростью подвижная фаза. По мере продвижения подвижной фазы с растворенными в ней веществами за определенный промежуток времени на колонке происходит разделение смеси. Чем больше сродство компонента к неподвижной фазе и чем меньше к подвижной, тем медленее он движется по колонке и тем дольше он в ней удерживается.
ЖХ можно разделить на 3 класса:
· адсорбционную;
· ионообменную;
· гельпроникающую (ситовая)
Адсорбционная хроматография:
Разделение веществ происходит за счет их различной способности адсорбироваться и десорбироваться с поверхности сорбента.
Этим методом можно анализировать липиды, сахара, ПАВ, витамины, гормоны
Адсорбционную жидкостную хроматографию можно разделить на 2 вида:
Ø Нормально-фазовая хроматография:
хроматография на полярном сорбенте (SiO2, Al2O3, силикагель) с неполярным элюентом (гексан, гептан). Хорошо разделяются неполярные соединения (бензол, насыщенные углеводороды)
Удерживание веществ растет с увеличением их полярности. Элюирующая способность подвижной фазы увеличивается с ростом ее полярности.
Ø Обращенно-фазовая хроматография:
хроматография на неполярном сорбенте (силикагель) с полярным элюентом (спирт, ацетон). Хорошо разделяются полярные соединения (спирты, витамины, антидепрессанты, наркотики)
Удерживание веществ растет с увеличением их гидрофобности (неполярности). Чем больше содержание органического растворителя, тем выше элюирующая способность подвижной фазы.
Ионообменная хроматография:
Молекулы веществ смеси, диссоциированные в растворе на катионы и анионы, разделяются при движении через сорбент (катионит или анионит) за счет различной силы взаимодействия определяемых ионов с ионными группами сорбента.
Основное применение ИОХ:
· анализ анионов в технологии и экологии;
· анализ металлов в технологии, воде, окружающей среде;
· ядерная физика, анализ белков и нуклеиновых кислот.
Гель-проникающая хроматография (ситовая, эксклюзионная):
Молекулы веществ разделяются по размеру за счет их разной способности проникать в поры неподвижной фазы. При этом первыми из колонки выходят наиболее крупные молекулы, способные проникать в минимальное число пор неподвижной фазы, а последними выходят вещества с малыми размерами молекул.
Подвижная фаза (элюент)
Подвижная фаза в жидкостной хроматографии выполняет двоякую функцию:
1) обеспечивает перенос десорбированных молекул по колонке;
2) регулирует константы равновесия, а, следовательно, и удерживание в результате
взаимодействия с неподвижной фазой (сорбируясь на поверхности) и с
молекулами разделяемых веществ.
Подвижная фаза, прежде всего, должна растворять разделяемые компоненты. Основными характеристиками подвижных фаз являются ее элюирующая способность и селективность.
Элюирующая способность ПФ – это ее способность вступать в межмолекулярные взаимодействия с разделяемыми соединениями и группами на поверхности сорбента.Эти взаимодействия способствуют десорбции разделяемых соединений, более быстрому перемещению хроматографических зон.
Селективность подвижных фаз связана с их способностью к специфическим взаимодействиям с сорбатами, определяемыми их структурными признаками.
Подвижная фаза может состоять из одного растворителя, часто из двух, при необходимости – из трех и более. Состав подвижной фазы указывают как объемное соотношение входящих в нее растворителей.
Растворитель один подобрать трудно. Зато смесь растворителей очень увеличивает гибкость ВЭЖХ. Принцип составления таких смесей прост. Необходимо взять два индивидуальных растворителя, один из которых имеет заведомо недостаточную элюирующую силу, другой — заведомо избыточную. Из этих двух растворителей можно приготовить множество различных подвижных фаз. Часть из них будет обязательно обладать подходящей элюирующей силой
В нормально-фазовой хроматографии обычно применяются жидкие углеводороды (гексан, циклогексан, гептан) и другие относительно неполярные растворители с небольшими добавками полярных органических соединений, которые регулируют элюирующую силу подвижной фазы.
В обращено-фазовой хроматографии в качестве подвижной фазы используется вода или водно-органические смеси. Органическими добавками обычно служат полярные органические растворители (ацетонитрил и метанол).
Неподвижная фаза (сорбент)
Неподвижная фаза – твердое пористое вещество, которое помещают в металлическую трубку – колонку
Основной материал для сорбентов в ЖАХ — силикагель. Он механически прочен, обладает значительной пористостью, что дает большую обменную емкость при небольших размерах колонки.
Силикагель может быть модифицирован различными химическими группами, привитыми к поверхности ( С-18, CN, NH2, SO3H ), что позволяет использовать сорбенты на его основе для разделения самых различных классов соединений.
Основной недостаток силикагеля — малая химическая стойкость при рН<2 и рН>9
В качестве сорбентов обычно применяются:
- силикагель, оксид алюминия, используются в нормально-фазовой хроматографии. Механизм удерживания в данном случае – обычно адсорбция;
- силикагель, смолы или полимеры с привитыми кислотными или основными группами. Область применения – ионообменная и ионная хроматография;
- силикагель или полимеры с заданным распределением размеров пор (эксклюзионная хроматография);
- химически модифицированные сорбенты (сорбенты с привитыми фазами), приготовленные чаще всего на основе силикагеля. Механизм удерживания ‑ адсорбция или распределение между подвижной и неподвижной фазами. Область применения зависит от типа привитых функциональных групп. Некоторые типы сорбентов могут использоваться как в обращенной, так и в нормально фазовой хроматографии;
Высокая эффективность разделения обеспечивается высокой площадью поверхности частиц сорбента (которая является следствием их микроскопических размеров и наличия пор), а также равномерностью состава сорбента и плотной и равномерной его упаковкой.
Преимущество проявительного (элюентного) анализа (проба вводится вместе с элюентом):
-дает наиболее полное разделение, поскольку зоны сорбатов разделены зонами элюента;
-сорбент непрерывно регенерируется;
-параметры удерживания хорошо воспроизводимы.
86. ВЭЖХ. Характеристика элюирующей способности растворителей в методе ВЭЖХ. Факторы, влияющие на выбор подвижной фазы при хроматографировании. Способы изменения состава подвижной фазы при хроматографировании.
Основными характеристиками подвижных фаз являются их элюирующая сила и се-лективность.
Один из элюентов способен смыть с колонки лишь слабосвязанные сорбаты, другие же вызывают десорбцию почти любых молекул. Молекулы подвижной фазы могут взаимо-действовать с молекулами разделяемых веществ. При этом образуются ассоциаты, которые взаимодействуют с неподвижной фазой, сила этого взаимодействия отличается от силы взаимодействия молекул пробы. В результате сорбция может стать более или менее прочной. С другой стороны, молекулы подвижной фазы могут конкурировать на поверхности сорбента с молекулами разделяемых соединений, вытесняя последние с активных центров и способствуя смещению равновесия в сторону десорбции.
Для характеристики влияния подвижной фазы на удерживание используют понятие элюирующей силы
Элюирующая сила подвижной фазы — это ее свойство вступать в такие межмолекулярные взаимодействия с компонентами системы, которые способствуют десорбции разделяемых соединений, более быстрому перемещению хроматографических зон.
Растворитель один подобрать трудно. Зато смесь растворителей очень увеличивает гибкость ВЭЖХ. Принцип составления таких смесей прост. Необходимо взять два индивидуальных растворителя, один из которых имеет заведомо недостаточную элюирующую силу, другой — заведомо избыточную. Из этих двух растворителей можно приготовить множество различных подвижных фаз. Часть из них будет обязательно обладать подходящей элюирующей силой.
В составе почти любой ПФ можно найти компонент сорбционно менее активный, выполняющий преимущественно транспортную функцию (растворитель А), и сорбционно активный, который служит для регулирования равновесия (растворитель В).
Роль одного и того же компонента в различных подвижных фазах и в зависимости от характера НФ, может быть различной. Например, в ПФ гексан-хлороформ последнее соединение выступает в качестве растворителя В (активного), а в системе хлороформ — метанол оно же играет роль растворителя А (транспортного).
С целью повышения селективности часто используют подвижные фазы более сложного состава, чем бинарные. Во многих случаях это приводит к улучшению разделения.
87. ВЭЖХ. Неподвижные фазы для ВЭЖХ. Способы получения, их характеристика. Теории, определяющие механизм разделения в обращенно-фазовой ВЭЖХ.
Неподвижная фаза (сорбент)
Основной материал для сорбентов в ЖАХ — силикагель. Он механически прочен, обладает значительной пористостью, что дает большую обменную емкость при небольших размерах колонки.
Силикагель может быть модифицирован различными химическими группами, привитыми к поверхности ( С-18, CN, NH2, SO3H ), что позволяет использовать сорбенты на его основе для разделения самых различных классов соединений.
Основной недостаток силикагеля — малая химическая стойкость при рН<2 и рН>9
В качестве сорбентов обычно применяются:
- силикагель, оксид алюминия, используются в нормально-фазовой хроматографии. Механизм удерживания в данном случае – обычно адсорбция;
- силикагель, смолы или полимеры с привитыми кислотными или основными группами. Область применения – ионообменная и ионная хроматография;
- силикагель или полимеры с заданным распределением размеров пор (эксклюзионная хроматография);
- химически модифицированные сорбенты (сорбенты с привитыми фазами), приготовленные чаще всего на основе силикагеля. Механизм удерживания ‑ адсорбция или распределение между подвижной и неподвижной фазами. Область применения зависит от типа привитых функциональных групп. Некоторые типы сорбентов могут использоваться как в обращенной, так и в нормально фазовой хроматографии;
Высокая эффективность разделения обеспечивается высокой площадью поверхности частиц сорбента (которая является следствием их микроскопических размеров и наличия пор), а также равномерностью состава сорбента и плотной и равномерной его упаковкой.
Разделение в обращенно-газовой ВЭЖХ
Причиной сорбции в обращенно-фазовой хроматографии служит сильное притяжение полярных молекул растворителя одна к другой, как бы “прижимающее” растворенные менее полярные молекулы к неполярной поверхности (для полярных подвижных фаз, в особенности содержащих воду, характерно сильное кулоновское взаимодействие и образование водородных связей между молекулами растворителей)
88. ВЭЖХ. Основные процессы взаимодействия в колонке. Влияние температуры на хроматографический процесс в методе ВЭЖХ. Основные причины размывания хроматографического пика. Кинетическая теория хроматографии. Уравнение Ван-Деемтера, величины и их вклад при изменении условий хроматографирования в методе ВЭЖХ.
При повышении температуры вязкость растворителя снижается, а следовательно и давление при хроматографировании → изменяется удерживание вещества
Вещества вводятся в колонку в виде узкой зоны, которая по мере ее движения с подвижной фазой по колонке становится все шире, т.е. размывается в результате диффузионных процессов
Мерой этого размывания в колонке является высота, эквивалентная теоретической тарелке (Н)
Установлено, что размывание полосы в хроматографической колонке обусловлено тремя причинами: наличием вихревой диффузии, молекулярной диффузии и сопротивления массопередаче.
Кинетическая теория хроматографии объясняет размывание хроматографических пиков этими тремя независимыми процессами, вклад каждого из которых описывается уравнением Ван-Деемтера:
H=A B/U CU, где
А – неравномерность движения потока элюента (вихревая диффузия),
B/U – молекулярная (продольная) диффузия миграция в подвижной фазе из участка с большей концентрацией в участок с меньшей,
CU – учитывает сопротивление массопереносу при непрерывном переходе вещества из подвижной фазы в неподвижную и обратно. Характеризует скорость распределения вещества между двумя фазами.
Таким образом, размывание в колонке уменьшается и эффективность повышается, когда применяется более мелкий сорбент, более равномерный по составу (узкая фракция), более плотно и равномерно упакованный в колонке, при использовании более тонких слоев неподвижной фазы, менее вязких подвижных фаз и оптимальных скоростей потока
89. ВЭЖХ. Метод теоретических тарелок в хроматографии. Теоретически минимальное значение для метода ВЭЖХ. Сравнение колоночного и микроколоночного вариантов. Эффективность хроматографической колонки. Способы повышения эффективности в методе ВЭЖХ. Состав и свойства сорбента в ВЭЖХ.
Теоретическая тарелка – это условный участок хроматографической колонки, в пределах которого устанавливается равновесие вещества, распределенного между подвижной и неподвижной фазами (одна теоретическая тарелка отвечает однократному распределению между фазами).Чем больше теоретических тарелок в колонке, чем большее число раз устанавливается равновесие, тем эффективнее колонка.
Эффективность колонки характеризуется числом теоретических тарелок и высотой, эквивалентной теоретической тарелке (ВЭТТ). Число теоретических тарелок,обозначаемое как n, является основной характеристикой и рассчитывается по формулам:
и где μ и μ0,5 соответственно ширина пика у основания и на середине высоты, измеренные в единицах времени удерживания.
Высота, эквивалентная теоретической тарелке (Н), рассчитывается по формуле, где L – длина колонки, и выражается в см.
Эффективность колонки тем выше, чем меньше высота, эквивалентная теоретической тарелке, и больше число теоретических тарелок.
Эффективность хроматографической колонки оценивают по нахождению экспериментальных значений хроматографических параметров:
-фактор удерживания (емкости) (k);
-коэффициент селективности (a);
-степень размывания хроматографического пика;
-разрешение (Rs) Разделение двух соседних пиков
Фактор удерживания (коэффициент емкости) – показывает во сколько раз дольше вещество пребывает в неподвижной фазе, чем в подвижной
Коэффициент селективности – мера относительного удерживания или относительной подвижности двух разделяемых сорбатов
Эффективность колонки в обычной ВЭЖХ (250*4,6 мм) (т.т.) 15 000-20 000
Эффективность колонки в микроколоночной ВЭЖХ (75*2 мм) (т.т.) 5 000-6 000
Неподвижная фаза (сорбент)
Основной материал для сорбентов в ЖАХ — силикагель. Он механически прочен, обладает значительной пористостью, что дает большую обменную емкость при небольших размерах колонки.
Силикагель может быть модифицирован различными химическими группами, привитыми к поверхности ( С-18, CN, NH2, SO3H ), что позволяет использовать сорбенты на его основе для разделения самых различных классов соединений.
Основной недостаток силикагеля — малая химическая стойкость при рН<2 и рН>9
В качестве сорбентов обычно применяются:
- силикагель, оксид алюминия, используются в нормально-фазовой хроматографии. Механизм удерживания в данном случае – обычно адсорбция;
- силикагель, смолы или полимеры с привитыми кислотными или основными группами. Область применения – ионообменная и ионная хроматография;
- силикагель или полимеры с заданным распределением размеров пор (эксклюзионная хроматография);
- химически модифицированные сорбенты (сорбенты с привитыми фазами), приготовленные чаще всего на основе силикагеля. Механизм удерживания ‑ адсорбция или распределение между подвижной и неподвижной фазами. Область применения зависит от типа привитых функциональных групп. Некоторые типы сорбентов могут использоваться как в обращенной, так и в нормально фазовой хроматографии;
Высокая эффективность разделения обеспечивается высокой площадью поверхности частиц сорбента (которая является следствием их микроскопических размеров и наличия пор), а также равномерностью состава сорбента и плотной и равномерной его упаковкой
90. ВЭЖХ. Основные узлы жидкостного хроматографа, их назначение. Модели хроматографов, используемых в практике в современных условиях. Насосы, используемые в жидкостных хроматографах, характеристика их работы.
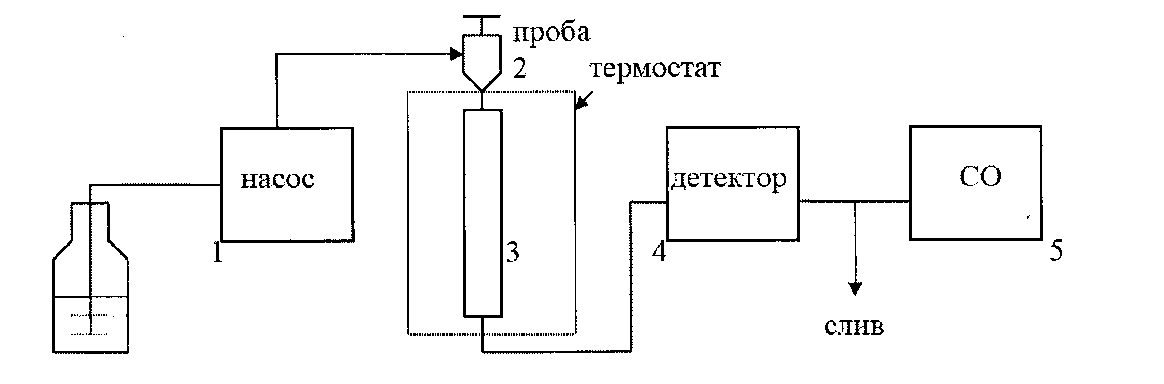
В состав любого хроматографа входят пять обязательных составных частей:
а) насос для подачи подвижной фазы через колонку;
б) дозатор для введения пробы в колонку;
в) разделительная колонка — сердце хроматографа♥
г) детектор — устройство для получения аналитического сигнала, пропорционального
концентрации компонента;
д) система обработки — преобразователь аналитического сигнала в форму удобную для
восприят
Колонка plot с пористым слоем сорбента hp-plot al2o3 kcl, 50 м x 0,32 мм х 8,00 мкм, 19091p-k15
Колонка PLOT с пористым слоем сорбента HP-PLOT Al2O3 KCl
Особенности:
— Наименее «полярная» окисно-алюминиевая фаза
— Оксид алюминия деактивирован KCl
— Стандартный выбор колонки для анализа лёгких углеводородов — изомеров
углеводородов от С
1
до С
8
— Низкое удерживание олефинов по сравнению с соответствующими
парафинами
— Отлично подходит для количественного определения диенов, особенно
пропадиена и бутадиена из потоков этилена и пропилена
— Рекомендуемая фаза для многих методов ASTM
— Предпочтительна дезактивация оксида алюминия KCl
Правила выбора колонки для газовой
хроматографии
Сформулируйте аналитическую задачу:
— Какие соединения и компоненты разделяемой смеси анализируются?
— В какой среде проводится анализ?
— Какой диапазон концентраций определяемых компонентов и какая точность
определения необходимы?
— Какие химико-физические характеристики разделяемых компонентов
являются
наиболее важными?
— Какое количество анализов необходимо проводить в день, неделю, месяц?
Для формулирования аналитической
задачи используйте:
— Методические указания национальных и международных стандартов таких
как
ГОСТ, ГОСТ Р ИСО, ASTM, EN, ISO, UOP, IP
— Методические указания отраслевых стандартов таких как СанПиН, ПНД Ф,
РД, МКХА, ОСТ, СТО Газпром, МУК, АЮВ, МВИ, М-, М-МВИ, М СПЭК, МР и др.
— Методические указания нормативных документов федеральных учреждений
России, таких как Ростехнадзор, Роспотребнадзор, Минздравсоцразвития,
ЭКЦ МВД и методические указания аналитических и исследовательских
центров
— Данные реферируемых научных журналов. Для быстрого обзора
электронные библиотеки такие как Pubmed, e-Library, Agilent
Searchable Chromatogram Library, ACD/Chromatography Applications
Database, Liquid Chromatography Data Base Search
— Руководство по выбору колонок Agilent J&W GC Column
Selection Guide
Выберите неподвижную фазу
При выборе типа неподвижной фазы начинайте с анализа примеров
применения колонок для разделения аналогичных или близких по составу
смесей в каталоге Agilent Technologies. Необходимо помнить, что с
увеличением полярности фазы времена выхода полярных соединений
увеличиваются, а максимальная рабочая температура колонок уменьшается.
Выберите внутренний диаметр колонок ID
(mm)
Чем больше внутренний диаметр колонки, тем большее ее емкость, и
тем большую массу разделяемых веществ можно ввести, не опасаясь
перегрузки колонки (без ухудшения разрешения). Так, для колонок с
внутренним диаметром 0.2 мм (толщина фазы 0.25 мкм) емкость составляет
35-70 нг, а для колонок 0.53 мм — 1000-2000 нг. Однако увеличение
внутреннего диаметра приводит к потере разрешения при прочих равных
условиях. Колонки с большим внутренним диаметром требуют меньшего
давления газа-носителя на входе для получения той же линейной скорости
потока.
Выберите длину колонки Length (m)
Увеличение длины колонки приводит к увеличению её разрешения
(пропорционально квадратному корню длины) но увеличивает также время
анализа и требуемое давление на входе в колонку. Необходимо выбрать
оптимальное разрешение не ухудшая производительность всей системы.
Длина колонки также влияет на рост её стоимости.
Выберите толщину неподвижной фазы Film
(μm)
Увеличение толщины пленки неподвижной фазы увеличивает времена
удерживания анализируемых соединений, повышает емкость колонок и
разрешение для легколетучих соединений, при этом увеличение толщины
пленки способствует о снижению рабочей температуры колонки и большему
уносу фазу, что особенно наглядно проявляется в конце хроматограммы,
когда унос фазы при высоких температурах приводит к подъему нулевой
линии.